
Le stampanti 3D hanno fatto notizia a causa del loro utilizzo negli ospedali per stampare tamponi e DPI. In questo articolo parliamo delle implicazioni normative della stampa 3D per queste applicazioni.
Comprendere la classificazione dei dispositivi
Negli Stati Uniti, l’Agenzia per gli alimenti e i medicinali (FDA) individua tre classi di dispositivi. La classificazione è determinata dagli usi previsti e dalle indicazioni per l’uso. Le differenze di classificazione possono voler dire che per il dispositivo in questione è richiesta l’autorizzazione o l’approvazione prima dell’immissione sul mercato (PMA) e controlli di produzione su più livelli.
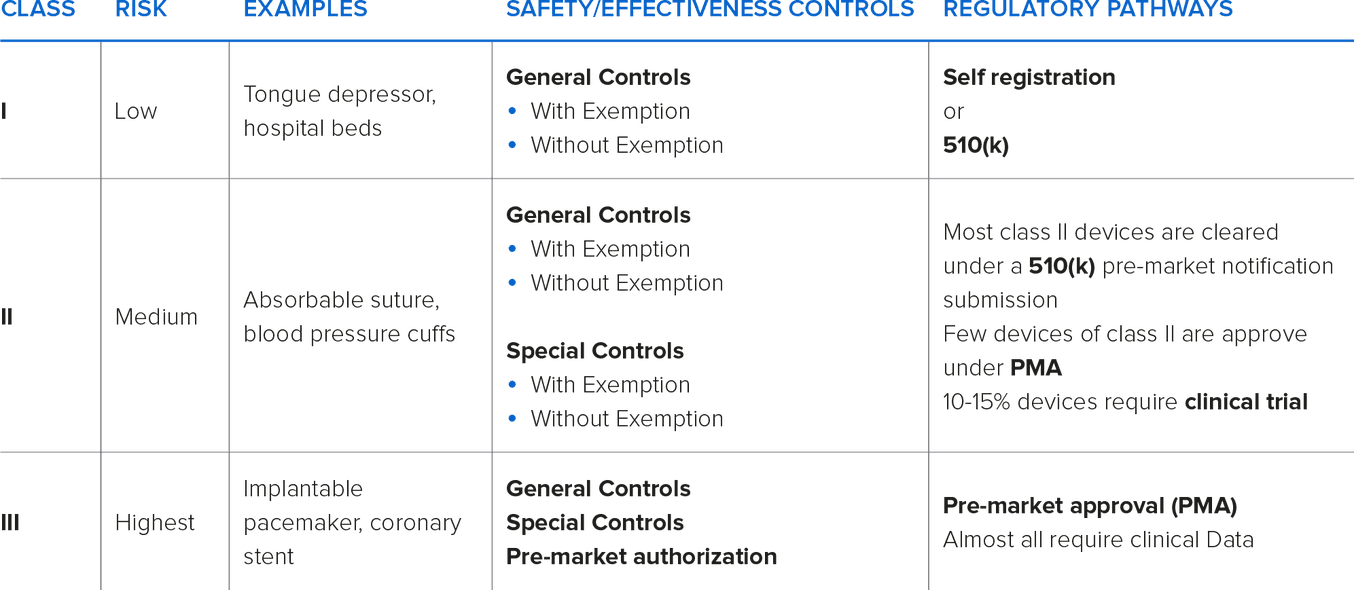
Solitamente, con l’aumentare del grado di classificazione, si rendono necessarie autorizzazioni o approvazioni prima dell’immissione sul mercato.
I dispositivi di Classe I e Classe II possono essere dichiarati “Esenti” o “Non Esenti”. I dispositivi di Classe I e II dichiarati esenti sono esonerati dalla notifica prima dell’immissione sul mercato. Alcuni dispositivi di Classe I possono essere esonerati anche dai requisiti che soddisfano le Norme di Buona Fabbricazione (NBF).
La maggior parte dei dispositivi di Classe I e alcuni di Classe II non sono soggetti ai requisiti per la notifica prima dell’immissione sul mercato, ma sono soggetti ad alcune limitazioni. Un dispositivo è dichiarato non soggetto ai requisiti previsti dal modulo 510(k) se l’Agenzia per gli alimenti e i medicinali degli Stati Uniti (FDA) determina che l’autorizzazione 510(k) non è richiesta per garantire la sicurezza e l’efficacia del dispositivo. Il programma 510(k) è usato per ottenere l’approvazione dimostrando la sostanziale equivalenza del dispositivo rispetto a un altro già sul mercato, chiamato “predicate device” (dispositivo di confronto). La sostanziale equivalenza di un dispositivo rispetto al dispositivo di confronto è determinata da: usi previsti e caratteristiche tecniche identiche oppure usi previsti identici e caratteristiche tecniche differenti ma equivalenza in termini di sicurezza ed efficacia; documentazione inviata all’Agenzia per gli alimenti e i medicinali degli Stati Uniti (FDA) a dimostrazione che il suddetto dispositivo è sicuro ed efficace tanto quanto il dispositivo già legalmente presente sul mercato.
I dispositivi di Classe III sono dispositivi ad alto rischio e possono essere usati per “sostenere o preservare la vita”. Questi dispositivi devono superare tutti i controlli dell’Agenzia per gli alimenti e i medicinali degli Stati Uniti (FDA) e delle Norme di Buona Fabbricazione (NBF) per ottenere l’approvazione alla loro immissione sul mercato.
Il produttore del dispositivo deve assumersi la responsabilità di classificare il proprio dispositivo in modo adeguato e di eseguire i controlli previsti. Per aiutare con la classificazione, la FDA dispone di una banca dati di dispositivi immessi sul mercato e delle classificazioni loro associate.
I professionisti abilitati come medici, dentisti e oculisti che producono o modificano i dispositivi esclusivamente per usi legati alla loro professione di solito non sono tenuti a soddisfare i requisiti di presentazione prima dell’immissione sul mercato. Dato che la FDA non ha l’autorità di regolamentare il lavoro di medici o infermieri, i dispositivi su misura per i pazienti che vengono stampati in ambito medico vanno oltre il campo d’applicazione della tradizionale regolamentazione dei dispositivi medici, essendo considerati “esercizio della professione medica.” Gli specialisti hanno la responsabilità di stabilire in quali circostanze questo approccio è appropriato.
Registrazione di dispositivi medici e biocompatibilità
Le resine BioMed di Formlabs sono materiali funzionali applicabili a una vasta gamma di usi in base alle esigenze di biocompatibilità e prestazioni. Dato che le applicazioni mediche sono di ampia portata e in costante evoluzione, la gamma di resine BioMed è progettata per soddisfare e superare i requisiti di una grande varietà di norme sanitarie e di sicurezza che permettono agli utenti di selezionare i materiali in base ai requisiti di progettazione dei loro dispositivi medici. Le resine BioMed, inoltre, sono conformi alle norme ISO 13485, che stabiliscono il controllo continuo della qualità, che potrebbe essere necessario per l’ottenimento da parte degli utenti di autorizzazioni o approvazioni per i dispositivi medici in produzione. Spesso, queste misure sono richieste per la presentazione prima dell’immissione sul mercato, ma non sono di per sé sufficienti a eliminare tutti gli ostacoli normativi legati alla registrazione di un dispositivo medico finito.
La documentazione di biocompatibilità è richiesta per l’invio dell’autorizzazione 510(k) e dell’approvazione prima dell’immissione sul mercato, dato che attesta se il dispositivo sarà compatibile con il sistema biologico a cui è destinato. Informazioni mancanti o insufficienti possono comportare notevoli ritardi nell’immissione del dispositivo sul mercato.
I requisiti dei test di biocompatibilità vanno determinati sulla base dell’uso previsto per il dispositivo (tipologia, area e durata dell’esposizione). Determinare i requisiti dei test all’inizio del processo di sviluppo concederà più margine di tempo per completare i test prima dell’invio. I requisiti vanno ricavati dal tipo di contatto previsto tra il dispositivo e il corpo umano. Il contatto è solitamente definito da tre diverse categorie:
-
Contatto diretto: il dispositivo entra in contatto fisico con i pazienti.
-
Contatto indiretto: il dispositivo entra in contatto fisico con un fluido, un gas o altro materiale che poi ha un contatto diretto con i pazienti.
-
Nessun contatto: non si verifica nessun tipo di contatto, né diretto né indiretto, con i pazienti, quindi non è soggetto ai requisiti di biocompatibilità.
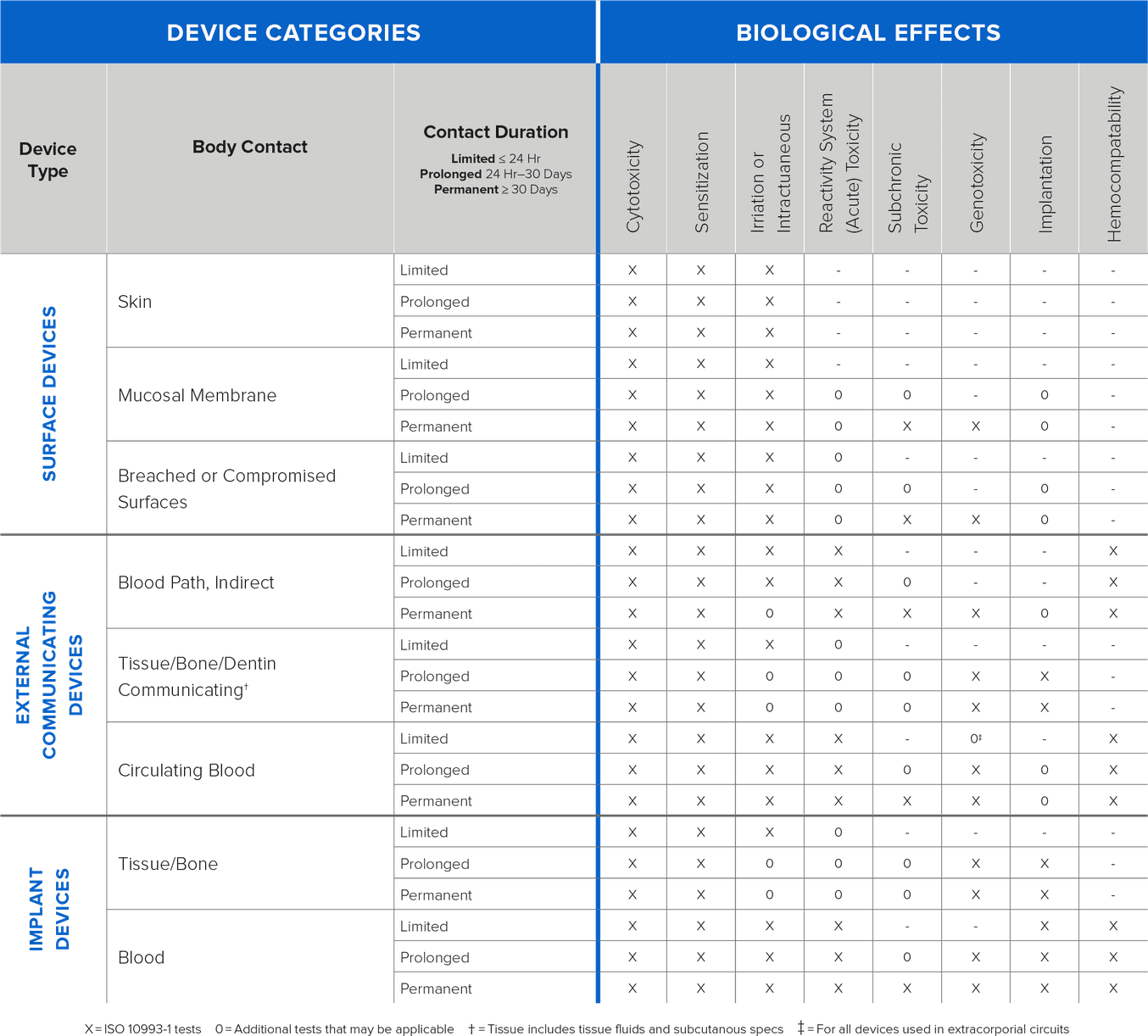
{Informazioni sulle tipologie di test basati sul contatto e sulla durata}
Formlabs esegue test di biocompatibilità basati sulle norme ISO 10993, ISO 18562 e ISO 7405 e pubblica le informazioni riguardanti le resine in questione. Dato che i test vengono condotti su campioni stampati standardizzati, i produttori che utilizzano design con geometrie complesse e altre modifiche devono convalidare la biocompatibilità dei dispositivi finiti per gli usi previsti.
Sistemi di gestione della qualità
Le attrezzature Formlabs per la stampa e la post-elaborazione vengono usate esclusivamente per produrre dispositivi medici, ma non sono esse stesse dispositivi medici. Quindi, non necessitano di autorizzazione o approvazione. I controlli di produzione devono essere determinati e applicati all’uso della stampante e dell’attrezzatura di post-elaborazione per assicurare una produzione uniforme e sicura di dispositivi con l’attrezzatura Formlabs.
È responsabilità del produttore assicurarsi che il dispositivo, compresa la verifica di design, materiali e post-elaborazione, sia sicuro ed efficace per l’uso a cui è destinato. È inoltre fondamentale che la struttura in cui vengono prodotti i dispositivi sia adatta alla produzione. Il resto dell’articolo si focalizzerà su ulteriori considerazioni rispetto al processo di stampa 3D dei dispositivi medici.
Stabilire il controllo del processo: prima di passare alla produzione, è importante convalidare il tuo processo. La capacità del tuo apparecchio deve risultare idonea alla riproduzione di parti identiche alle linee guida di design. Considera le seguenti domande:
-
Quali fattori possono influenzare il risultato della tua stampa? Questi fattori possono includere impostazioni dell’apparecchio/ambiente, design della piattaforma di stampa ecc.
-
Come puoi controllare i fattori sopra citati per garantire che ogni parte sia prodotta secondo specifiche predeterminate?
Questa pianificazione prestampa ti aiuterà a sostenere la riuscita del lavoro.
Mantenere il controllo del processo: considera in che modo puoi controllare i fattori decisivi per le tue stampe. Eccoti un paio di esempi di come potresti riuscirci:
-
Crea una Procedura Operativa Standard (POS) per stampare ed eseguire la post-elaborazione. Questa procedura può includere:
1. File STL standard
2. Controlli prestampa standard (preparazione della resina, livellamento della stampante ecc.)
3. Attività post-stampa standard (tempi minimi dalla fine della stampa al primo lavaggio, tempi/posizioni standard di lavaggio e polimerizzazione ecc.)
-
Considera le attività periodiche pianificate necessarie per mantenere la tua attrezzatura e il processo in condizioni adeguate. Ciò può includere la sostituzione del serbatoio, la sostituzione di liquido di lavaggio/alcool isopropilico o i controlli della stampante. Documenta i tuoi piani e l’esecuzione delle attività pianificate.
Controlli di qualità: stabilisci il modo in cui verificherai che parti completate sono accettabili per l’uso. Per farlo dovrai scegliere un metodo e la frequenza con cui effettuare l’ispezione.
-
Esempio di metodo: confronto con una specifica, come la misurazione della lunghezza in confronto alla lunghezza specificata, e il confronto con uno standard, come il confronto di un campione con una parte già convalidata come corretta per determinare se la lunghezza è accettabile.
-
Esempio di frequenza: ispezionare alcune parti incluse in ogni stampa, tutte le parti incluse in determinate stampe (ad es. una stampa ogni cinque), tutte le parti incluse in ogni stampa.
Non dimenticare di pianificare il controllo di qualità di ogni elemento essenziale del prodotto (dimensionale, meccanico, funzionale). Elementi diversi possono avere frequenze e metodi di controllo qualità diversi.
Aspettarsi l’inaspettato: anche i processi che si conoscono meglio possono comportarsi in modo inaspettato. Prepara un piano per poter reagire in modo appropriato e proteggere i pazienti da prodotti potenzialmente pericolosi. Alcuni processi comuni per reagire in modo appropriato:
- Crea un registro dei lotti per segmentare e identificare ogni stampa in modo univoco. Per ciascuna stampa, documenta informazioni come il lotto di resina, il serbatoio, eventuali errori riscontrati durante la stampa, l’attrezzatura di post-elaborazione, ecc. Se dovessi riscontrare un problema sconosciuto, queste informazioni potrebbero rivelarsi cruciali per determinare la causa principale e separare in modo appropriato i dispositivi funzionanti da quelli difettosi.
-
Prepara un piano di risposta. Considera cosa potrebbe andare storto durante il processo di stampa e stabilisci come reagire:
1. Cosa farai se alcune parti sulla piattaforma di stampa dovessero risultare difettose? Scarterai tutte le parti o solo quelle visibilmente non accettabili?
2. Se durante la stampa dovesse essere individuato un errore, procederai con il riavvio della stampa o la annullerai?
Avere un piano di cosa fare in casi del genere ti aiuterà ad agire in modo uniforme.


