
3D printers have been in the news with respect to hospitals printing test swabs and PPE. In this post, we talk about the regulatory implications of 3D printing for these applications.
Understand Device Classifications
In the United States, the (Food and Drug Administration) FDA designates three classifications of devices. Classification is determined by intended use and indications for use. Classification differences may mean that the device requires a premarket clearance or approval (PMA) information and varying degrees of manufacturing controls.
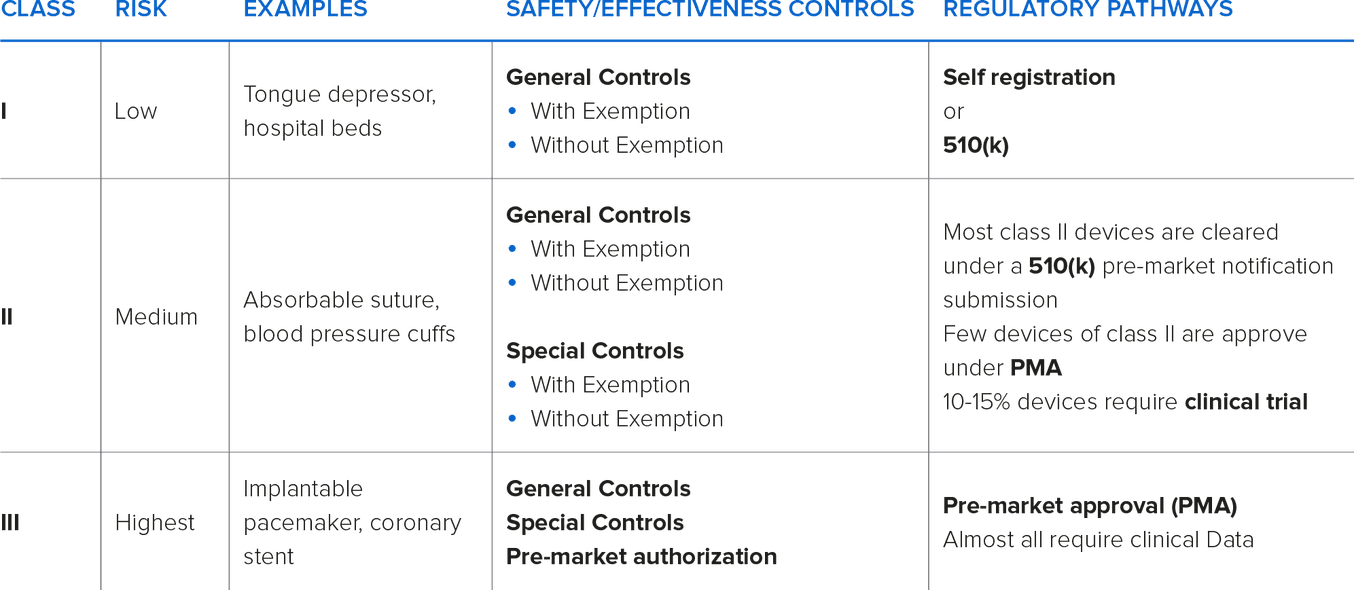
Typically, as the classification increases, premarket clearances or approvals are necessary.
Class I and Class II devices can be considered either ‘Exempt’ or ‘Non-Exempt’. Exempt Class I & II devices are exempt from premarket notification. Some Class I devices may also be exempt from Good Manufacturing Practices (GMPs) requirements.
Most Class I and some Class II devices are exempt from premarket notification requirements, subject to certain limitations. A device may be exempt from 510(k) requirements if the FDA determines that a 510(k) is not required to provide reasonable assurance of safety and effectiveness for the device. The 510(k) program is used to achieve clearance through demonstrating substantial equivalency to a device already on the market, called a predicate device. A device is substantially equivalent if, in comparison to a predicate it: has the same intended use as the predicate; and has the same technological characteristics as the predicate; or has the same intended use as the predicate; and has different technological characteristics and does not raise different questions of safety and effectiveness; and the information submitted to the FDA demonstrates that the device is as safe and effective as the legally marketed device.
Class III devices are the highest risk devices that can be used to “support or sustain life”. The devices require full FDA PMA and GMP controls.
The manufacturer of the device assumes the responsibility of properly classifying their device and implementing the appropriate controls. For help with classification, the FDA maintains a database of marketed devices and their associated classifications.
Licensed practitioners, including physicians, dentists, and optometrists, who manufacture or otherwise alter devices solely for use in their practice, are typically exempt from premarket submission requirements. Since the FDA does not have the authority to regulate a physician's or nurse's practice, patient-specific devices printed in a medical setting are beyond the scope of traditional medical device regulations, as they’re considered “the practice of medicine.” The clinician maintains the responsibility to determine where this approach is appropriate.
Medical Device Registration & Biocompatibility
Formlabs BioMed Resins are functional materials applicable for a wide range of uses based on biocompatibility and performance needs. As healthcare applications are far-reaching and ever-evolving, the BioMed Resin portfolio is designed to meet/exceed an inclusive variety of health and safety standards that allow users to select materials based on their medical device design requirements. BioMed Resins are also ISO 13485 compliant which provides the continual quality assurance that may be necessary for a user to achieve clearance or approval for the medical device being manufactured. Often, these measures are required in a premarket submission, but is not enough in itself to clear all the regulatory hurdles of registering a finished medical device.
Biocompatibility information is required for 510(k) and PMA submissions, as it proves whether the device will be compatible in the biological system in which it is intended. Missing or inadequate information can lead to significant delays in bringing the device to market.
Biocompatibility testing requirements should be determined based on intended use of the device (type, area, and duration of exposure). Determining testing requirements early in the development process will allow ample time to complete testing prior to submission and should stem from the expected contact between the device and the human body. Contact is typically defined by three different categories:
-
Direct Contact: Comes into physical contact with the patient.
-
Indirect Contact: Comes into physical contact with a fluid, gas, or other material that then has direct contact with the patient.
-
No Contact: Does not have direct or indirect contact with the patient and is therefore exempt from biocompatibility requirements.
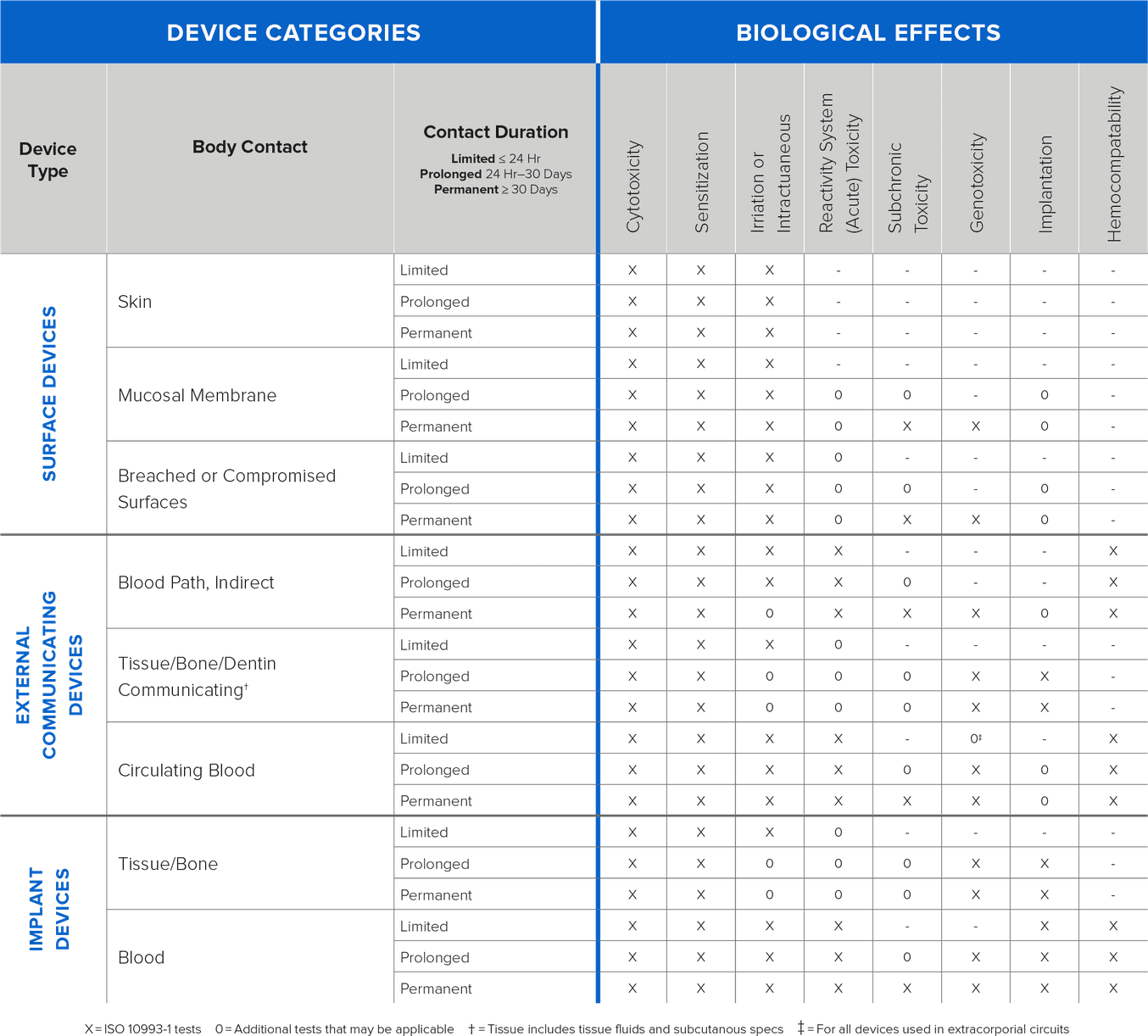
{Information on types of testing based on contact and duration}
Formlabs conducts biocompatibility testing based on ISO 10993, ISO 18562, and ISO 7405 standards and publishes the information on relevant resins. As the testing is conducted on standardized printed samples, manufacturers with complex design geometries, and other modifications must validate biocompatibility of the finished devices for their intended use.
Quality Management System
Formlabs printing and post-processing equipment are only used to produce medical devices and are not medical devices themselves. Therefore, they do not require clearance or approval. Manufacturing controls need to be determined and applied to the use of the printer and post-processing equipment to ensure consistent, safe production of devices on Formlabs equipment.
It is a manufacturer’s responsibility to ensure the device, including the validation of design, materials, and post-processing, is safe and effective for use. It is also imperative that the facility that the devices are produced in is acceptable for production. The remainder of this article will focus on additional considerations for the process of 3D printing medical devices.
Establish process control: Prior to production, it is important to validate your process. This should consist of qualifying your machine’s capability to reproduce parts identical to the design specifications. Consider the following questions:
-
What factors may affect the outcome of your print? These factors may include machine setup/environment, design of the build platform, etc.
-
How can you control the above factors to ensure each part is manufactured according to predetermined specifications?
This preprint planning will help you sustain a successful operation.
Maintain process control: Consider the ways in which you can control factors critical to your prints. Below are a few examples of how this may be accomplished:
-
Create a Standard Operating Procedure (SOP) for printing and post-processing. This may include the following:
1. Standard .STL files
2. Standard pre-print checks (resin preparation, printer leveling, etc)
3. Standard post-printing activities (minimum time from print finish to first wash, standard wash and cure times/positions, etc.)
-
Consider planned periodic activities required to keep your equipment and process in a capable condition. This may include tank replacement, IPA/wash fluid replacement, or printer checks. Document your plans and the execution of the planned activities.
Quality checks: Determine how you will ensure completed parts are acceptable for use. To do this, you will need to choose a method and a frequency for inspection.
-
Example method: Comparison to a specification, such as measuring the length and comparing to the specified length, and comparison to a standard, such as comparing a sample part to a known good part to determine if the length is acceptable.
-
Example frequencies: Inspecting a few parts per build, all parts on every few builds, all parts every build.
Remember to have a quality control (QC) plan for each essential element of the product (dimensional, mechanical, functional). Different elements can have different QC frequencies and methods.
Expect the unexpected: Even the most understood processes often behave in unexpected ways. Make plans to allow you to respond appropriately and protect patients from potentially unsafe products. A few common processes to allow appropriate response:
- Create a batch record to segment and uniquely identify each build. For each build, document information such as the resin batch, tank, any errors encountered in printing, post-processing equipment, etc. If you encounter an unfamiliar issue, this information may be crucial in determining root cause and appropriately bracketing the good devices from the bad.
-
Have a reaction plan. Consider the things that can go wrong in the printing process and determine how you will react:
1. If a few parts on your build platform fail, what will you do? Will you discard all parts, or only those that are visibly not acceptable?
2. If your print encounters an error, will you restart or abort the print?
Having a plan of how you will react to common scenarios will help you remain consistent in your actions.